介绍
扩增子测序是分析环境微生物的常见手段,通常使用的是16s rRNA片段。16srRNA分析主要有质控、去冗余、聚类OTU、去嵌合体、生成OTU表和物种注释等步骤。更多知识分享请到 https://zouhua.top/。
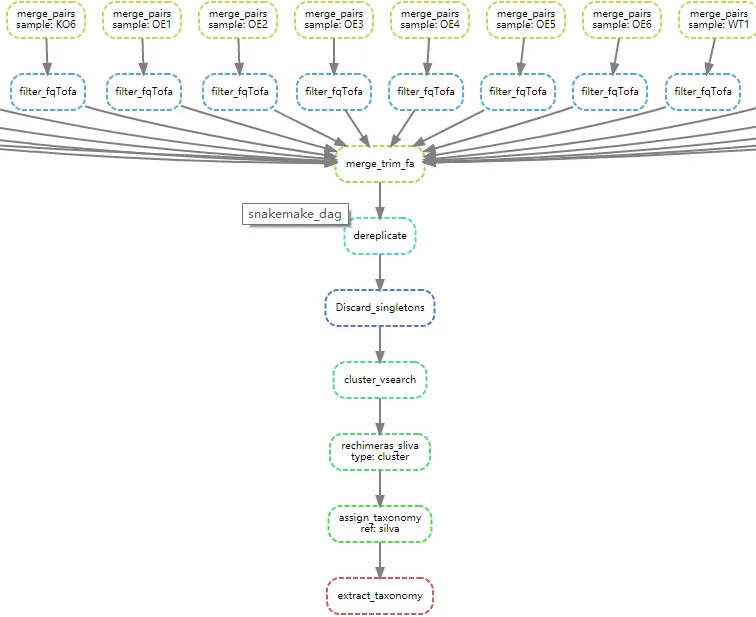
先看看前期数据处理的可视化图。
数据
18份来自宏基因组公众号的双端16s rRNA原始下机数据。
| sample | fq1 | fq2 |
|---|---|---|
| KO1 | data/KO1_1.fq.gz | data/KO1_2.fq.gz |
| KO2 | data/KO2_1.fq.gz | data/KO2_2.fq.gz |
| KO3 | data/KO3_1.fq.gz | data/KO3_2.fq.gz |
| KO4 | data/KO4_1.fq.gz | data/KO4_2.fq.gz |
| KO5 | data/KO5_1.fq.gz | data/KO5_2.fq.gz |
| KO6 | data/KO6_1.fq.gz | data/KO6_2.fq.gz |
| OE1 | data/OE1_1.fq.gz | data/OE1_2.fq.gz |
| OE2 | data/OE2_1.fq.gz | data/OE2_2.fq.gz |
| OE3 | data/OE3_1.fq.gz | data/OE3_2.fq.gz |
| OE4 | data/OE4_1.fq.gz | data/OE4_2.fq.gz |
| OE5 | data/OE5_1.fq.gz | data/OE5_2.fq.gz |
| OE6 | data/OE6_1.fq.gz | data/OE6_2.fq.gz |
| WT1 | data/WT1_1.fq.gz | data/WT1_2.fq.gz |
| WT2 | data/WT2_1.fq.gz | data/WT2_2.fq.gz |
| WT3 | data/WT3_1.fq.gz | data/WT3_2.fq.gz |
| WT4 | data/WT4_1.fq.gz | data/WT4_2.fq.gz |
| WT5 | data/WT5_1.fq.gz | data/WT5_2.fq.gz |
| WT6 | data/WT6_1.fq.gz | data/WT6_2.fq.gz |
步骤
- 质控
- 去冗余
- 聚类OTU
- 去嵌合体
- 生成OTU表
- 物种注释
import os
import sys
import shutil
import pandas as pd
configfile: "config.yaml"
samples = pd.read_csv(config["samples"], sep="\t", index_col=["sample"])
rule all:
input:
expand("{taxonomy}/taxonomy.{{ref}}.{{type}}.{res}",
taxonomy=config["results"]["taxonomy"],
ref=["sliva", "RDP"],
type=["cluster","unoise3"],
res=["biom","mothur","txt"])
include: "rules/00.fastqc.snk"
include: "rules/01.trim.snk"
include: "rules/02.deredundancy.snk"
include: "rules/03.clusterOTU.snk"
include: "rules/04.rechimeras.snk"
include: "rules/05.OTUtable.snk"
include: "rules/06.assign_taxonomy.snk"
#include: "rules/07.picrust.snk" # 这一步还没有实现
质控
质控包括剔除质量低的reads和切除带有barcodes的接头等
检查reads质量情况
- 设置通配符函数,匹配样本名字,wildcards是snakemake的通配符函数。
- fastqc和multiqc软件可以生成可视化的reads质量分布情况等的网页版文件。
- config是snakemake内调用配置文件的参数。
- fastqc和multiqc的结果可以确定质控过程reads应该保留的长度。
去冗余
- 移除出现频率低的reads,这些reads可能是测序错误引起的,minuniquesize参数越大,相对而言计算量越小,正常建议设置成2,只去除单次出现的序列。
- Discard_singletons排序后再去除单次出现的序列。
rule dereplicate:
input:
os.path.join(config["results"]["trim"], "summary_trimmed.fa")
output:
temp(os.path.join(config["results"]["deredundancy"], "deredundancy.fa"))
params:
name = config["params"]["deredundancy"]["name"],
mins = config["params"]["deredundancy"]["mins"]
log:
os.path.join(config["logs"], "02.deredundancy.log")
shell:
'''
vsearch --derep_fulllength {input} --output {output} --relabel {params.name} \
--minuniquesize {params.mins} --sizeout 2>{log}
'''
rule Discard_singletons:
input:
os.path.join(config["results"]["deredundancy"], "deredundancy.fa")
output:
os.path.join(config["results"]["deredundancy"], "sorted.fa")
params:
size = config["params"]["deredundancy"]["size"]
log:
os.path.join(config["logs"], "02.sorted.log")
shell:
'''
vsearch --sortbysize {input} --output {output} --minsize {params.size} 2>{log}
'''
聚类OTU
- 根据序列相似性聚类(一般阈值通常为97%)能有效避免测序错误,将聚类后的序列集合称为可操作分类单元(operational taxonomic units OTUs),每一个OTU代表一类物种。常用的方法有uparse、unoise3等,但这些方法没有考虑因单碱基多态性而存在的序列多样性,于是deblur和DADA2算法被开放出来针对聚类和单碱基多态性。这里我们暂时用uparse和unoise3算法。
rule cluster_vsearch:
input:
os.path.join(config["results"]["deredundancy"], "sorted.fa")
output:
fa = os.path.join(config["results"]["clusterOTU"], "OTU.cluster.fa"),
biom = os.path.join(config["results"]["clusterOTU"], "OTU.cluster.biom"),
mothur = os.path.join(config["results"]["clusterOTU"], "OTU.cluster.mothur"),
tab = os.path.join(config["results"]["clusterOTU"], "OTU.cluster.txt"),
uc = os.path.join(config["results"]["clusterOTU"], "OTU.cluster.uc")
params:
identity = config["params"]["clusterOTU"]["identity"],
threads = config["params"]["clusterOTU"]["threads"],
name = config["params"]["clusterOTU"]["name"]
log:
os.path.join(config["logs"], "03.clusterOTU.vsearch.log")
shell:
'''
vsearch --threads {params.threads} --cluster_fast {input} --id {params.identity} \
--centroids {output.fa} --biomout {output.biom} \
--mothur_shared_out {output.mothur} --otutabout {output.tab} \
--relabel {params.name} --uc {output.uc} 2>{log}
'''
rule cluster_uparse:
input:
os.path.join(config["results"]["deredundancy"], "sorted.fa")
output:
os.path.join(config["results"]["clusterOTU"], "OTU.uparse.fa")
params:
name = config["params"]["clusterOTU"]["name"]
log:
os.path.join(config["logs"], "03.clusterOTU.uparse.log")
shell:
'''
usearch11 -cluster_OTUs {input} -otus {output} -relabel {params.name} 2>{log}
'''
rule cluster_unoise3:
input:
os.path.join(config["results"]["clusterOTU"], "OTU.uparse.fa")
output:
os.path.join(config["results"]["clusterOTU"], "OTU.unoise3.fa")
log:
os.path.join(config["logs"], "03.clusterOTU.unoise3.log")
shell:
'''
usearch11 -unoise3 {input} -zotus {output} 2>{log}
'''
去嵌合体
- 嵌合体的产生是因为PCR过程可能某两端DNA序列连接在一起而扩增。
- 去嵌合体的原理是将序列比对到对应的参考数据库,16s rRNA的参考数据库主要有RDP、GreenGenes和sliva,其中常用的是RDP和sliva。
- 去嵌合体后保留的序列即为代表序列。
rule rechimeras_sliva:
input:
expand("{cluster}/OTU.{{type}}.fa",
cluster=config["results"]["clusterOTU"],
type=["cluster","unoise3"])
output:
borderline = os.path.join(config["results"]["rechimeras"], "chimeric.{type}.borderline"),
nonchimeras = os.path.join(config["results"]["rechimeras"], "OTU.rechimera_silva.{type}.fa"),
chimeras = os.path.join(config["results"]["rechimeras"], "chimeric_silva.{type}.sequence")
params:
db = config["params"]["rechimeras"]["silva"]
log:
os.path.join(config["logs"], "04.OTU.rechimera_silva.{type}.log")
shell:
'''
vsearch --uchime_ref {input} --db {params.db} --borderline {output.borderline} \
--chimeras {output.chimeras} --nonchimeras {output.nonchimeras} 2>{log}
'''
rule rechimeras_RDP:
input:
expand("{cluster}/OTU.{{type}}.fa",
cluster=config["results"]["clusterOTU"],
type=["cluster","unoise3"])
output:
os.path.join(config["results"]["rechimeras"], "OTU.rechimera_RDP.{type}.fa")
params:
db = config["params"]["rechimeras"]["rdp"]
log:
os.path.join(config["logs"], "04.OTU.rechimera_RDP.{type}.log")
shell:
'''
usearch11 -uchime2_ref {input} -db {params.db} -chimeras {output} -strand plus -mode balanced 2>{log}
'''
生成OTU表
- 聚类生成的OTU比对到去嵌合体后的OTU得到OTU表。
- 设置比对阈值 identity: 97%。
rule make_otutab_vsearch:
input:
otu_ref = expand("{rechimeras}/OTU.rechimera_{{ref}}.{{type}}.fa",
rechimeras=config["results"]["rechimeras"],
ref=["sliva", "RDP"],
type=["cluster","unoise3"]),
trim_fa = os.path.join(config["results"]["trim"], "summary_trimmed.fa")
output:
aln = os.path.join(config["results"]["OTUtable"], "OTU_table.{ref}.{type}.shr.aln"),
biom = os.path.join(config["results"]["OTUtable"], "OTU_table.{ref}.{type}.biom"),
mothur = os.path.join(config["results"]["OTUtable"], "OTU_table.{ref}.{type}.mothur"),
uc = os.path.join(config["results"]["OTUtable"], "OTU_table.{ref}.{type}.uc"),
txt = os.path.join(config["results"]["OTUtable"], "OTU_table.{ref}.{type}.txt")
params:
identity = config["params"]["OTUtable"]["identity"],
threads = config["params"]["OTUtable"]["threads"]
log:
os.path.join(config["logs"], "05.OTU_table.{ref}.{type}.log")
shell:
'''
vsearch --usearch_global {input.trim_fa} --db {input.otu_ref} --id {params.identity}\
--strand plus --threads {params.threads} --alnout {output.aln} --biomout {output.biom} \
--mothur_shared_out {output.mothur} --uc {output.uc} --otutabout {output.txt} 2>{log}
'''
rule OTU_tight_clusters:
input:
otu_ref = expand("{rechimeras}/OTU.rechimera_{{ref}}.{{type}}.fa",
rechimeras=config["results"]["OTUtable"],
ref=["sliva", "RDP"],
type=["cluster","unoise3"])
output:
fa = os.path.join(config["results"]["OTUtable"], "OTU.rechimera.{ref}.{type}_new.fa"),
uc = os.path.join(config["results"]["OTUtable"], "OTU.rechimera.{ref}.{type}_hits.uc")
params:
identity = config["params"]["OTUtable"]["identity"],
accepts = config["params"]["OTUtable"]["accepts"],
rejects = config["params"]["OTUtable"]["rejects"]
log:
os.path.join(config["logs"], "05.OTU_table.{ref}.{type}.log")
shell:
'''
usearch11 -cluster_fast {input.otu_ref} -id {params.identity} \
-maxaccepts {params.accepts} -maxrejects {params.rejects} \
-top_hit_only -uc {output.uc} -centroids {output.fa} 2>{log}
'''
物种注释
- 代表序列比对到16s rRNA参考数据库。
- 控制比对identity参数。
rule assign_taxonomy:
input:
otu_ref = expand("{rechimeras}/OTU.rechimera_{{ref}}.{{type}}.fa",
rechimeras=config["results"]["rechimeras"],
ref=["sliva", "RDP"],
type=["cluster","unoise3"])
output:
biom = os.path.join(config["results"]["taxonomy"], "taxonomy.{ref}.{type}.biom"),
mothur = os.path.join(config["results"]["taxonomy"], "taxonomy.{ref}.{type}.mothur"),
txt = os.path.join(config["results"]["taxonomy"], "taxonomy.{ref}.{type}.txt")
params:
identity = config["params"]["taxonomy"]["identity"],
threads = config["params"]["taxonomy"]["threads"],
database = config["params"]["taxonomy"]["silva"]
log:
os.path.join(config["logs"], "06.taxonomy.{ref}.{type}.log")
shell:
'''
vsearch --usearch_global {input.otu_ref} --db {params.database} \
--biomout {output.biom} --mothur_shared_out {output.mothur} --otutabout {output.txt} \
--id {params.identity} --threads {params.threads} 2>{log}
'''